

Getting Started#
Installation#
You can install sgkit with pip. Python 3.11 or 3.12 is required:
$ pip install sgkit
Alternatively, you can use conda:
$ conda install -c conda-forge sgkit
Overview#
Sgkit is a general purpose toolkit for quantitative and population genetics. The primary goal of sgkit is to take advantage of powerful tools in the PyData ecosystem to facilitate interactive analysis of large-scale datasets. The main libraries we use are:
Data structures#
Sgkit uses Xarray Dataset objects to model genetic data.
Users are free to manipulate quantities within these objects as they see fit and a set of conventions for variable names,
dimensions and underlying data types is provided to aid in workflow standardization. The example below illustrates a
Dataset format that would result from an assay expressible as VCF,
PLINK or BGEN.
This is a guideline however, and a Dataset seen in practice might include many more or fewer variables and dimensions.
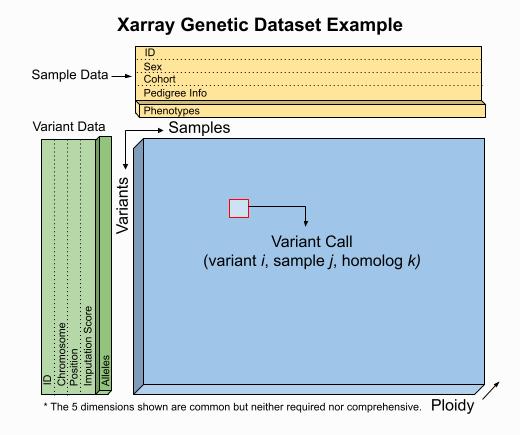
The worked examples below show how to access and visualize data like this using Xarray. They also demonstrate several of the sgkit conventions in place for representing common genetic quantities.
In [1]: import sgkit as sg
In [2]: ds = sg.simulate_genotype_call_dataset(n_variant=1000, n_sample=250, n_contig=23, missing_pct=.1)
In [3]: ds
Out[3]:
<xarray.Dataset> Size: 1MB
Dimensions: (contigs: 23, variants: 1000, alleles: 2, samples: 250,
ploidy: 2)
Dimensions without coordinates: contigs, variants, alleles, samples, ploidy
Data variables:
contig_id (contigs) <U2 184B '0' '1' '2' '3' ... '20' '21' '22'
variant_contig (variants) int64 8kB 0 0 0 0 0 0 0 ... 22 22 22 22 22 22
variant_position (variants) int64 8kB 0 1 2 3 4 5 6 ... 37 38 39 40 41 42
variant_allele (variants, alleles) |S1 2kB b'G' b'A' b'T' ... b'A' b'T'
sample_id (samples) <U4 4kB 'S0' 'S1' 'S2' ... 'S248' 'S249'
call_genotype (variants, samples, ploidy) int8 500kB 0 0 1 0 ... 0 0 1
call_genotype_mask (variants, samples, ploidy) bool 500kB False ... False
Attributes:
contigs: ['0', '1', '2', '3', '4', '5', '6', '7', '8', '9', '10', '11', ...
source: sgkit-unknown
The presence of a single-nucleotide variant (SNV) is indicated above by the call_genotype variable, which contains
an integer value corresponding to the index of the associated allele present (i.e. index into the variant_allele variable)
for a sample at a given genome coordinate and chromosome. Every sgkit variable has a set of fixed semantics like this. For more
information on this specific variable and any others, see Genetic variables.
Data subsets can be accessed as shown here, using the features described in Indexing and Selecting Xarray Data:
In [4]: import sgkit as sg
In [5]: ds = sg.simulate_genotype_call_dataset(n_variant=100, n_sample=50, n_contig=23, missing_pct=.1)
# Subset the entire dataset to the first 10 variants/samples
In [6]: ds.isel(variants=slice(10), samples=slice(10))
Out[6]:
<xarray.Dataset> Size: 884B
Dimensions: (contigs: 23, variants: 10, alleles: 2, samples: 10,
ploidy: 2)
Dimensions without coordinates: contigs, variants, alleles, samples, ploidy
Data variables:
contig_id (contigs) <U2 184B '0' '1' '2' '3' ... '20' '21' '22'
variant_contig (variants) int64 80B 0 0 0 0 0 1 1 1 1 1
variant_position (variants) int64 80B 0 1 2 3 4 0 1 2 3 4
variant_allele (variants, alleles) |S1 20B b'T' b'C' b'C' ... b'T' b'A'
sample_id (samples) <U3 120B 'S0' 'S1' 'S2' ... 'S7' 'S8' 'S9'
call_genotype (variants, samples, ploidy) int8 200B 0 0 1 0 ... 1 1 1
call_genotype_mask (variants, samples, ploidy) bool 200B False ... False
Attributes:
contigs: ['0', '1', '2', '3', '4', '5', '6', '7', '8', '9', '10', '11', ...
source: sgkit-unknown
# Subset to a specific set of variables
In [7]: ds[['variant_allele', 'call_genotype']]
Out[7]:
<xarray.Dataset> Size: 10kB
Dimensions: (variants: 100, alleles: 2, samples: 50, ploidy: 2)
Dimensions without coordinates: variants, alleles, samples, ploidy
Data variables:
variant_allele (variants, alleles) |S1 200B b'T' b'C' b'C' ... b'T' b'A'
call_genotype (variants, samples, ploidy) int8 10kB 0 0 1 0 1 ... 0 1 0 0
Attributes:
contigs: ['0', '1', '2', '3', '4', '5', '6', '7', '8', '9', '10', '11', ...
source: sgkit-unknown
# Extract a single variable
In [8]: ds.call_genotype[:3, :3]
Out[8]:
<xarray.DataArray 'call_genotype' (variants: 3, samples: 3, ploidy: 2)> Size: 18B
array([[[ 0, 0],
[ 1, 0],
[ 1, 0]],
[[ 1, 1],
[ 1, 0],
[ 1, -1]],
[[ 0, 1],
[ 1, 1],
[ 1, 1]]], dtype=int8)
Dimensions without coordinates: variants, samples, ploidy
Attributes:
mixed_ploidy: False
comment: Call genotype. Encoded as allele values (0 for the referen...
# Access the array underlying a single variable (this would return dask.array.Array if chunked)
In [9]: ds.call_genotype.data[:3, :3]
Out[9]:
array([[[ 0, 0],
[ 1, 0],
[ 1, 0]],
[[ 1, 1],
[ 1, 0],
[ 1, -1]],
[[ 0, 1],
[ 1, 1],
[ 1, 1]]], dtype=int8)
# Access the alleles corresponding to the calls for the first variant and sample
In [10]: allele_indexes = ds.call_genotype[0, 0]
In [11]: allele_indexes
Out[11]:
<xarray.DataArray 'call_genotype' (ploidy: 2)> Size: 2B
array([0, 0], dtype=int8)
Dimensions without coordinates: ploidy
Attributes:
mixed_ploidy: False
comment: Call genotype. Encoded as allele values (0 for the referen...
In [12]: ds.variant_allele[0, allele_indexes]
Out[12]:
<xarray.DataArray 'variant_allele' (ploidy: 2)> Size: 2B
array([b'T', b'T'], dtype='|S1')
Dimensions without coordinates: ploidy
Attributes:
comment: The possible alleles for the variant.
# Get a single item from an array as a Python scalar
In [13]: ds.sample_id.item(0)
Out[13]: 'S0'
Larger subsets of data can be visualized and/or summarized through various sgkit utilities as well as the Pandas/Xarray integration:
In [14]: import sgkit as sg
In [15]: ds = sg.simulate_genotype_call_dataset(n_variant=1000, n_sample=250, missing_pct=.1)
# Show genotype calls with domain-specific display logic
In [16]: sg.display_genotypes(ds, max_variants=8, max_samples=8)
Out[16]:
samples S0 S1 S2 S3 ... S246 S247 S248 S249
variants ...
0 0/0 1/0 1/0 0/1 ... 1/1 0/0 1/1 1/0
1 0/0 0/1 1/1 1/1 ... 0/1 1/0 0/. 0/0
2 1/1 0/1 1/0 0/1 ... 0/0 1/1 0/1 ./1
3 1/0 1/0 1/0 1/0 ... 1/1 1/0 0/1 1/.
... ... ... ... ... ... ... ... ... ...
996 0/. 1/1 1/1 0/0 ... ./. 0/0 1/0 ./0
997 0/0 ./0 1/0 1/1 ... 1/. 1/0 1/1 ./1
998 0/1 0/1 1/1 1/0 ... 0/0 0/0 0/1 ./1
999 1/0 0/1 1/0 1/1 ... 0/1 1/0 0/0 0/1
[1000 rows x 250 columns]
# A naive version of the above is also possible using only Xarray/Pandas and
# illustrates the flexibility that comes from being able to transition into
# and out of array/dataframe representations easily
In [17]: (ds.call_genotype[:5, :5].to_series()
....: .unstack().where(lambda df: df >= 0, None).fillna('.')
....: .astype(str).apply('/'.join, axis=1).unstack())
....:
Out[17]:
samples 0 1 2 3 4
variants
0 0.0/0 1.0/0 1.0/0 0.0/1 ./0
1 0.0/0 0.0/1 1.0/1 1.0/1 ./0
2 1.0/1 0.0/1 1.0/0 0.0/1 1.0/0
3 1.0/0 1.0/0 1.0/0 1.0/0 1.0/1
4 ./1 ./1 1.0/1 0.0/0 0.0/1
# Show call rate distribution for each variant using Pandas
In [18]: df = ~ds.call_genotype_mask.to_dataframe()
In [19]: df.head(5)
Out[19]:
call_genotype_mask
variants samples ploidy
0 0 0 True
1 True
1 0 True
1 True
2 0 True
In [20]: call_rates = df.groupby('variants').mean()
In [21]: call_rates
Out[21]:
call_genotype_mask
variants
0 0.898
1 0.910
2 0.926
3 0.894
4 0.892
... ...
995 0.892
996 0.906
997 0.900
998 0.900
999 0.910
[1000 rows x 1 columns]
In [22]: call_rates.plot(kind='hist', bins=24, title='Call Rate Distribution', figsize=(6, 3))
Out[22]: <Axes: title={'center': 'Call Rate Distribution'}, ylabel='Frequency'>
This last example alludes to representations of missing data that are explained further in Missing data.
Genetic methods#
Genetic methods in sgkit are nearly always applied to individual Dataset objects. For a full list of
available methods, see Methods.
In this example, the variant_stats method is applied to a dataset to compute a number of statistics
across samples for each individual variant:
In [23]: import sgkit as sg
In [24]: ds = sg.simulate_genotype_call_dataset(n_variant=100, n_sample=50, missing_pct=.1)
In [25]: sg.variant_stats(ds, merge=False)
Out[25]:
<xarray.Dataset> Size: 9kB
Dimensions: (variants: 100, alleles: 2)
Dimensions without coordinates: variants, alleles
Data variables:
variant_n_called (variants) int64 800B dask.array<chunksize=(100,), meta=np.ndarray>
variant_call_rate (variants) float64 800B dask.array<chunksize=(100,), meta=np.ndarray>
variant_n_het (variants) int64 800B dask.array<chunksize=(100,), meta=np.ndarray>
variant_n_hom_ref (variants) int64 800B dask.array<chunksize=(100,), meta=np.ndarray>
variant_n_hom_alt (variants) int64 800B dask.array<chunksize=(100,), meta=np.ndarray>
variant_n_non_ref (variants) int64 800B dask.array<chunksize=(100,), meta=np.ndarray>
variant_allele_count (variants, alleles) uint64 2kB dask.array<chunksize=(100, 2), meta=np.ndarray>
variant_allele_total (variants) int64 800B dask.array<chunksize=(100,), meta=np.ndarray>
variant_allele_frequency (variants, alleles) float64 2kB dask.array<chunksize=(100, 2), meta=np.ndarray>
There are two ways that the results of every function are handled – either they are merged with the provided dataset or they are returned in a separate dataset. See Dataset merge behavior for more details.
Missing data#
Missing data in sgkit is represented using a sentinel value within data arrays
(-1 in integer arrays and NaN in float arrays) as well as a companion boolean mask array
(True where data is missing). These sentinel values are handled transparently in
most sgkit functions and where this isn’t possible, limitations related to it are documented
along with potential workarounds.
This example demonstrates one such function where missing calls are ignored:
In [26]: import sgkit as sg
In [27]: ds = sg.simulate_genotype_call_dataset(n_variant=1, n_sample=4, n_ploidy=2, missing_pct=.3, seed=4)
In [28]: ds.call_genotype
Out[28]:
<xarray.DataArray 'call_genotype' (variants: 1, samples: 4, ploidy: 2)> Size: 8B
array([[[ 0, 0],
[ 1, 1],
[-1, 0],
[-1, -1]]], dtype=int8)
Dimensions without coordinates: variants, samples, ploidy
Attributes:
mixed_ploidy: False
comment: Call genotype. Encoded as allele values (0 for the referen...
# Here, you can see that the missing calls above are not included in the allele counts
In [29]: sg.count_variant_alleles(ds).variant_allele_count
Out[29]:
<xarray.DataArray 'variant_allele_count' (variants: 1, alleles: 2)> Size: 16B
dask.array<sum-aggregate, shape=(1, 2), dtype=uint64, chunksize=(1, 2), chunktype=numpy.ndarray>
Dimensions without coordinates: variants, alleles
Attributes:
comment: Variant allele counts. With shape (variants, alleles) and value...
A primary design goal in sgkit is to facilitate ad hoc analysis. There are many useful functions in the library but they are not enough on their own to accomplish many analyses. To that end, it is often helpful to be able to handle missing data in your own functions or exploratory summaries. Both the sentinel values and the boolean mask array help make this possible, where the sentinel values are typically more useful when implementing compiled operations and the boolean mask array is easier to use in a higher level API like Xarray or Numpy. Only advanced users would likely ever need to worry about compiling their own functions (see Custom Computations for more details). Using Xarray functions and the boolean mask is generally enough to accomplish most tasks, and this mask is often more efficient to operate on due to its high on-disk compression ratio. This example shows how it can be used in the context of doing something simple like counting heterozygous calls:
In [30]: import sgkit as sg
In [31]: import xarray as xr
In [32]: ds = sg.simulate_genotype_call_dataset(n_variant=1, n_sample=4, n_ploidy=2, missing_pct=.2, seed=2)
# This array contains the allele indexes called for a sample
In [33]: ds.call_genotype
Out[33]:
<xarray.DataArray 'call_genotype' (variants: 1, samples: 4, ploidy: 2)> Size: 8B
array([[[-1, 0],
[ 1, 1],
[ 1, 0],
[ 1, 1]]], dtype=int8)
Dimensions without coordinates: variants, samples, ploidy
Attributes:
mixed_ploidy: False
comment: Call genotype. Encoded as allele values (0 for the referen...
# This array represents only locations where the above calls are missing
In [34]: ds.call_genotype_mask
Out[34]:
<xarray.DataArray 'call_genotype_mask' (variants: 1, samples: 4, ploidy: 2)> Size: 8B
array([[[ True, False],
[False, False],
[False, False],
[False, False]]])
Dimensions without coordinates: variants, samples, ploidy
Attributes:
comment: A flag for each call indicating which values are missing.
# Determine which calls are heterozygous
In [35]: is_heterozygous = (ds.call_genotype[..., 0] != ds.call_genotype[..., 1])
In [36]: is_heterozygous
Out[36]:
<xarray.DataArray 'call_genotype' (variants: 1, samples: 4)> Size: 4B
array([[ True, False, True, False]])
Dimensions without coordinates: variants, samples
# Count the number of heterozygous samples for the lone variant
In [37]: is_heterozygous.sum().item(0)
Out[37]: 2
# This is almost correct except that the calls for the first sample aren't
# really heterozygous, one of them is just missing. Conditional logic like
# this can be used to isolate those values and replace them in the result:
In [38]: xr.where(ds.call_genotype_mask.any(dim='ploidy'), False, is_heterozygous).sum().item(0)
Out[38]: 1
# Now the result is correct -- only the third sample is heterozygous so the count should be 1.
# This how many sgkit functions handle missing data internally:
In [39]: sg.variant_stats(ds).variant_n_het.values.item(0)
Out[39]: 1
Windowing#
It is common to compute statistics in windows along the genome. Some Methods in sgkit are “windowing aware” and will compute values for windows defined in a dataset. If no windows are defined then the values will typically be computed for each variant. It is therefore important to define windows before computing statistics on a dataset.
Windows are intervals that span the variants dimension in a dataset, and they are defined
using the sgkit.window_by_variant(), sgkit.window_by_position(), and
sgkit.window_by_genome() functions.
The first function, sgkit.window_by_variant(), is the simplest and produces windows with
a fixed number of variants in each window, determined by the size argument. An
optional step argument may be provided to control the spacing between windows. By default,
it is the same as the size, giving contiguous windows.
The second function, sgkit.window_by_position(), produces windows whose size is measured
by genomic position (base pairs). See the API documentation for usage and examples.
The third function, sgkit.window_by_genome(), produces a single window spanning the whole
genome, which can be used to compute whole-genome statistics.
This example shows the effect of computing the diversity statistic: first with no windows defined, then with windows.
In [40]: import sgkit as sg
In [41]: import xarray as xr
In [42]: ds = sg.simulate_genotype_call_dataset(n_variant=100, n_sample=50)
# Define a single cohort for all samples
In [43]: ds["sample_cohort"] = xr.DataArray(np.full(ds.dims['samples'], 0), dims="samples")
# The diversity statistic is computed for every variant since no windows are defined
In [44]: sg.diversity(ds, merge=False)
Out[44]:
<xarray.Dataset> Size: 800B
Dimensions: (variants: 100, cohorts: 1)
Dimensions without coordinates: variants, cohorts
Data variables:
stat_diversity (variants, cohorts) float64 800B dask.array<chunksize=(100, 1), meta=np.ndarray>
# Define windows of size 20 variants. This creates a new dimension called `windows`, and
# some new variables for internal use.
In [45]: ds = sg.window_by_variant(ds, size=20)
# The diversity statistic is now computed for every window
In [46]: sg.diversity(ds, merge=False)
Out[46]:
<xarray.Dataset> Size: 40B
Dimensions: (windows: 5, cohorts: 1)
Dimensions without coordinates: windows, cohorts
Data variables:
stat_diversity (windows, cohorts) float64 40B dask.array<chunksize=(5, 1), meta=np.ndarray>
Cohorts#
During analysis we often want to be able to group samples into populations, and compute statistics based on these groups. Groups of samples are referred to as cohorts in sgkit.
Cohorts are defined by a mapping from samples to cohort index. The following example creates
a sample_cohort variable to group a dataset of ten samples into three cohorts. Note that first
value is -1, which means the corresponding sample is not in any of the three cohorts, and
will be ignored when computing cohort statistics.
In [47]: import sgkit as sg
In [48]: import xarray as xr
In [49]: ds = sg.simulate_genotype_call_dataset(n_variant=100, n_sample=10)
In [50]: ds["sample_cohort"] = xr.DataArray(np.array([-1, 0, 1, 1, 1, 1, 0, 2, 2, 2]), dims="samples")
Typically the sample_cohort variable is derived from a dataframe that has the sample/cohort
mapping.
Cohort-level statistics can have repeated cohorts dimensions. sgkit.Fst(), for example,
produces statistics for pairs of cohorts, which is represented as a variable with dimensions
(windows, cohorts_0, cohorts_1), making it possible to read off the value of the statistic
for any pair of cohorts.
It’s convenient to name cohorts, to avoid errors that can occur when using index values. This example shows how to give cohorts names.
In [51]: ds = sg.window_by_variant(ds, size=20)
In [52]: ds = sg.Fst(ds)
In [53]: cohort_names = ["Africa", "Asia", "Europe"]
In [54]: ds = ds.assign_coords({"cohorts_0": cohort_names, "cohorts_1": cohort_names})
In [55]: ds.stat_Fst.sel(cohorts_0="Africa", cohorts_1="Asia").values
Out[55]: array([ 0.00823045, -0.02510823, 0.04372041, 0.12240725, -0.11305475])
Methods that work with cohorts will, by default, operate over all cohorts at once. Sometimes however you might only want to run the computation for a subset of cohorts, in which case you can explicitly specify the cohorts when calling the function.
Chaining operations#
Method chaining is a common practice with Python data tools that improves code readability and reduces the probability of introducing accidental namespace collisions. Sgkit functions are compatible with this idiom by default and this example shows to use it in conjunction with Xarray and Pandas operations in a single pipeline:
In [56]: import sgkit as sg
In [57]: ds = sg.simulate_genotype_call_dataset(n_variant=100, n_sample=50, missing_pct=.1)
# Use `pipe` to apply a single sgkit function to a dataset
In [58]: ds_qc = ds.pipe(sg.variant_stats).drop_dims('samples')
In [59]: ds_qc
Out[59]:
<xarray.Dataset> Size: 11kB
Dimensions: (variants: 100, alleles: 2, contigs: 1)
Dimensions without coordinates: variants, alleles, contigs
Data variables: (12/13)
variant_n_called (variants) int64 800B dask.array<chunksize=(100,), meta=np.ndarray>
variant_call_rate (variants) float64 800B dask.array<chunksize=(100,), meta=np.ndarray>
variant_n_het (variants) int64 800B dask.array<chunksize=(100,), meta=np.ndarray>
variant_n_hom_ref (variants) int64 800B dask.array<chunksize=(100,), meta=np.ndarray>
variant_n_hom_alt (variants) int64 800B dask.array<chunksize=(100,), meta=np.ndarray>
variant_n_non_ref (variants) int64 800B dask.array<chunksize=(100,), meta=np.ndarray>
... ...
variant_allele_total (variants) int64 800B dask.array<chunksize=(100,), meta=np.ndarray>
variant_allele_frequency (variants, alleles) float64 2kB dask.array<chunksize=(100, 2), meta=np.ndarray>
contig_id (contigs) <U1 4B '0'
variant_contig (variants) int64 800B 0 0 0 0 0 0 ... 0 0 0 0 0 0
variant_position (variants) int64 800B 0 1 2 3 4 ... 95 96 97 98 99
variant_allele (variants, alleles) |S1 200B b'T' b'C' ... b'A'
Attributes:
contigs: ['0']
source: sgkit-unknown
# Show statistics for one of the arrays to be used as a filter
In [60]: ds_qc.variant_call_rate.to_series().describe()
Out[60]:
count 100.000000
mean 0.804800
std 0.063125
min 0.560000
25% 0.780000
50% 0.800000
75% 0.840000
max 0.960000
Name: variant_call_rate, dtype: float64
# Build a pipeline that filters on call rate and computes Fst between two cohorts
# for windows of size 20 variants
In [61]: (
....: ds
....: .pipe(sg.variant_stats).compute()
....: .pipe(lambda ds: ds.sel(variants=ds.variant_call_rate > .8))
....: .pipe(lambda ds: sg.window_by_variant(ds, size=20))
....: .assign(sample_cohort=np.repeat([0, 1], ds.dims['samples'] // 2))
....: .pipe(sg.Fst)
....: .stat_Fst.values
....: )
....:
Out[61]:
array([[[ nan, 0.00091611],
[ 0.00091611, nan]],
[[ nan, 0.00588756],
[ 0.00588756, nan]],
[[ nan, -0.00550994],
[-0.00550994, nan]]])
This is possible because sgkit functions nearly always take a Dataset as the first argument, create new
variables, and then merge these new variables into a copy of the provided dataset in the returned value.
See Dataset merge behavior for more details.
Chunked arrays#
Chunked arrays are required when working on large datasets. Libraries for managing chunked arrays such as Dask Array and Zarr make it possible to implement blockwise algorithms that operate on subsets of arrays (in parallel) without ever requiring them to fit entirely in memory.
By design, they behave almost identically to in-memory (typically Numpy) arrays within Xarray and can be interchanged freely when provided
to sgkit functions. The most notable difference in behavior though is that operations on chunked arrays are evaluated lazily.
This means that if an Xarray Dataset contains only chunked arrays, no actual computations will be performed
until one of the following occurs:
Dataset.compute is called
DataArray.compute is called
The
DataArray.valuesattribute is referencedIndividual dask arrays are retrieved through the
DataArray.dataattribute and forced to evaluate via Client.compute, dask.array.Array.compute or by coercing them to another array type (e.g. using np.asarray)
This example shows a few of these features:
In [62]: import sgkit as sg
In [63]: ds = sg.simulate_genotype_call_dataset(n_variant=100, n_sample=50, missing_pct=.1)
# Chunk our original in-memory dataset using a blocksize of 50 in all dimensions.
In [64]: ds = ds.chunk(chunks=50)
In [65]: ds
Out[65]:
<xarray.Dataset> Size: 22kB
Dimensions: (contigs: 1, variants: 100, alleles: 2, samples: 50,
ploidy: 2)
Dimensions without coordinates: contigs, variants, alleles, samples, ploidy
Data variables:
contig_id (contigs) <U1 4B dask.array<chunksize=(1,), meta=np.ndarray>
variant_contig (variants) int64 800B dask.array<chunksize=(50,), meta=np.ndarray>
variant_position (variants) int64 800B dask.array<chunksize=(50,), meta=np.ndarray>
variant_allele (variants, alleles) |S1 200B dask.array<chunksize=(50, 2), meta=np.ndarray>
sample_id (samples) <U3 600B dask.array<chunksize=(50,), meta=np.ndarray>
call_genotype (variants, samples, ploidy) int8 10kB dask.array<chunksize=(50, 50, 2), meta=np.ndarray>
call_genotype_mask (variants, samples, ploidy) bool 10kB dask.array<chunksize=(50, 50, 2), meta=np.ndarray>
Attributes:
contigs: ['0']
source: sgkit-unknown
# Show the chunked array representing base pair position
In [66]: ds.variant_position
Out[66]:
<xarray.DataArray 'variant_position' (variants: 100)> Size: 800B
dask.array<xarray-variant_position, shape=(100,), dtype=int64, chunksize=(50,), chunktype=numpy.ndarray>
Dimensions without coordinates: variants
Attributes:
comment: The reference position of the variant.
# Call compute via the dask.array API
In [67]: ds.variant_position.data.compute()[:5]
Out[67]: array([0, 1, 2, 3, 4])
# Coerce to numpy via Xarray
In [68]: ds.variant_position.values[:5]
Out[68]: array([0, 1, 2, 3, 4])
# Compute without unboxing from xarray.DataArray
In [69]: ds.variant_position.compute()[:5]
Out[69]:
<xarray.DataArray 'variant_position' (variants: 5)> Size: 40B
array([0, 1, 2, 3, 4])
Dimensions without coordinates: variants
Attributes:
comment: The reference position of the variant.
Unlike this simplified example, real datasets often contain a mixture of chunked and unchunked arrays. Sgkit
will often load smaller arrays directly into memory while leaving large arrays chunked as a trade-off between
convenience and resource usage. This can always be modified by users though and sgkit functions that operate
on a Dataset should work regardless of the underlying array backend.
See Parallel computing with Dask in Xarray for more examples and information, as well as the Dask tutorials on delayed array execution and lazy execution in Dask graphs.